Abstract and Introduction
Abstract
Parkinson's disease is the second most common neurodegenerative disease and yet the early pathophysiological events of the condition and sequences of dysfunction remain unclear. The loss of dopaminergic neurons and reduced levels of striatal dopamine are descriptions used interchangeably as underlying the motor deficits in Parkinson's disease. However, decades of research suggest that dopamine release deficits in Parkinson's disease do not occur only after cell death, but that there is dysfunction or dysregulation of axonal dopamine release before cell loss. Here we review the evidence for dopamine release deficits prior to neurodegeneration in Parkinson's disease, drawn from a large and emerging range of Parkinson's disease models, and the mechanisms by which these release deficits occur. The evidence indicates that impaired dopamine release can result from disruption to a diverse range of Parkinson's disease-associated genetic and molecular disturbances, and can be considered as a potential pathophysiological hallmark of Parkinson's disease.
Introduction
Parkinson's disease is one of the most common movement disorders, with debilitating motor symptoms associated with age-related progressive neurodegeneration. Although ~10% of Parkinson's disease cases can be linked to inherited monogenic mutations, collectively referred to as familial Parkinson's disease, most cases, which are referred to as idiopathic or sporadic, have undetermined origin.[1] The subpopulation of dopamine (DA)-producing neurons of the substantia nigra pars compacta (SNpc) in the midbrain are preferentially affected (Figure 1).[2,3] SNpc neurons project to the dorsal striatum forming the nigrostriatal pathway and contribute to basal ganglia circuits involved in action selection, modulation and learning. As hallmarks of Parkinson's disease, the death of dopaminergic neurons (DANs) and reduction of striatal DA are frequently referred to interchangeably in reports of Parkinson's disease studies. However, much work in the last decade indicates that DA deficits may not result solely from cell death. A large body of work has demonstrated that in many models of Parkinson's disease, deficits in DA release from nigrostriatal neurons are present without, or before, neurodegeneration (Figure 1).
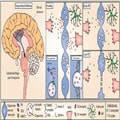
Figure 1.
DA release in the nigrostriatal pathway and simplified working hypothesis for Parkinson's disease progression. DAN cell bodies reside in the SNpc and project extensive axonal arbourizations to the dorsal striatum where they release DA (left). DA release occurs at active axonal varicosities and acts on D1 and D2 receptors on striatal cells (middle). In Parkinson's disease, the nigrostriatal pathway is most affected and results in progressively decreased DA release (top right), and eventually axon degeneration and cell death (bottom right) impairing downstream signalling and movement modulation. Cellular dysfunction and altered striatal DA release precede cell death. DA release is modulated by neighbouring cell types including astrocytes, which can contribute to DA release defects observed in Parkinson's disease models, such as by downregulation of GAT. Radiotracers used to image dopaminergic dysfunction in living human brains include 11C-raclopride that binds available D2 receptors and 18F-DOPA, which is a substrate for DOPA decarboxylase (DDC).
Animal models and, more recently, induced pluripotent stem cell (iPSC)-derived human neuronal models of monogenic Parkinson's disease, have been useful for understanding disease pathogenesis and functional changes in DANs that might contribute to Parkinson's disease.[4] Beyond changes to DA release, these changes include defects in autophagy, intracellular trafficking, mitochondria function and calcium homeostasis.[5–7] However, there is no unifying hypothesis or single convergent mechanism explaining the onset of Parkinson's disease to connect early cellular deficits to late-stage Parkinson's disease motor symptoms. In this review, we discuss DA release defects in Parkinson's disease models as an early and major pathological feature of the disease.
The Neurobiology of Dopamine Release
DA release is mediated by Ca2+ and Rab3-interacting molecule-dependent vesicular exocytosis from active zone-like sites.[8] In contrast to the conventional synapse, released DA is thought to act beyond individual synapses at extrasynaptic receptors on many cells and can also be released from the cell body and dendrites through somatodendritic release.[9] DA release is highly regulated through a variety of mechanisms, including DA synthesis, cell and vesicular uptake, action potential propagation and local neuromodulatory receptors.[10] DA is synthesized in the cytoplasm in consecutive reactions by the rate-limiting enzyme tyrosine hydroxylase (TH) and DOPA decarboxylase (DDC), coupled with its loading into synaptic vesicles (SVs) by vesicular monoamine transporter 2 (VMAT2) (Figure 1). The mechanisms through which DA vesicles are handled remain incompletely defined but might differ from many seen at other synapse types. For example, the synaptic molecular machinery for exocytosis from axons involves at least priming and release site scaffolding by Rab3-interacting molecule, munc13 and liprin-α, but might differ from other synapses in requirements for many other active zone proteins.[11] The biogenesis/recycling pathway of DA SVs involves adaptor protein3 (AP-3) rather than AP-2 that regenerates many other SVs.[12]
DA release dynamics in vivo are thought to be determined by a combination of different DAN firing rates (tonic low frequency pacemaking, sustained increases in firing rate and short intermittent or 'phasic' bursts at high frequencies), as well as by a range of mechanisms operating on DA axons that can locally gate action potential propagation, DA release probability and its dynamics during activity.[10,13] DA acts through D1- or D2-type receptors on recipient cells that probably include all striatal cells. Following its release, DA is primarily taken back up by DANs via the DA uptake transporter (DAT). It can also be degraded by monoamine oxidase B or catechol-O-methyltransferase.
SNpc DANs have distinctive axonal arbours formed by extensive branching of long unmyelinated axons[14] that bear exceptionally large numbers of release sites (>10[5]).[15] Their propagation of action potentials has been proposed to pose extremely high energetic demands.[16] These factors are suggested to contribute to their preferential vulnerability to degeneration in Parkinson's disease.[16] Early synaptic dysfunction in Parkinson's disease has long been reported as a key feature in both Parkinson's disease models and human patients.[17] Dopaminergic axons are affected early in Parkinson's disease, with synaptic decay preceding neuron death.[18,19] This concept is not unique to Parkinson's disease; impaired synaptic activity and function, referred to as synaptopathy, is observed in a wide variety of neurodegenerative diseases, including Huntington's, Alzheimer's and motor neuron disease.[18,20] Although many neurotransmitter pathways are affected in Parkinson's disease, and DA deficits are frequently considered a relatively late but characteristic pathology of the disease, a substantial body of work suggests DA dysfunction is an early cardinal feature.
Evidence for Early Synaptic Dysfunction and Dopamine Release Defects in Patients With Parkinson's Disease
Much evidence indicates that nigrostriatal degeneration begins in axons,[17] but is it preceded by DA release defects? Subjects who are at risk for Parkinson's disease, such as non-manifesting carriers of Parkinson's disease-associated mutations, asymptomatic family members or individuals with prodromal symptoms, represent the best opportunity to study preclinical Parkinson's disease and therefore to elucidate systems of early dysfunction.[21] Brain imaging studies in humans in vivo suggest presynaptic defects precede symptom onset in Parkinson's disease.[17,19,22,23] PET imaging of asymptomatic family members revealed presynaptic dopaminergic dysfunction, with reduced 18F-DOPA uptake, a measure of presynaptic dopaminergic integrity or DA handling, in individuals who later went on to develop Parkinson's disease.[24]18F-DOPA uptake was also significantly reduced in non-manifesting twins that were discordant for Parkinson's disease.[25,26] Of these twin pairs, several asymptomatic monozygotic cotwins with abnormal baseline scans later went on to develop clinical Parkinson's disease.[26] Similar findings were observed in familial Parkinson's disease-associated asymptomatic mutation carriers.[23,27,28] Two studies demonstrated that 18F-DOPA uptake was reduced in asymptomatic carriers with a single parkin (PRKN) mutation allele when compared to a control cohort.[23,28] A 5-year follow-up study suggested that although subclinical reductions of striatal 18F-DOPA uptake are frequent in single parkin mutation carriers, the rate of disease progression appeared very slow, although confirmation with a larger and longer-term longitudinal follow-up study, would be required.[29] Asymptomatic individuals heterozygous for PTEN-induced kinase 1 (PINK1) mutations also demonstrated a significant reduction in 18F-DOPA uptake when compared to a control cohort.[30] Studies demonstrate that in asymptomatic Y1699C and R1441C leucine-rich repeat kinase 2 (LRRK2) mutation carriers, 18F-DOPA uptake was normal, despite evidence for impaired DA function with abnormal DAT binding.[27,31] A longitudinal study that followed asymptomatic LRRK2 G2019S carriers who went on to convert to Parkinson's disease by the time of a 4-year re-evaluation revealed a lower striatal DAT binding at baseline than nonconverters.[32] Brain imaging of patients with prodromal Parkinson's disease, including hyposmic and REM sleep behaviour disorder cohorts, has also revealed dopaminergic dysfunction.[21,22] Combined, these studies suggest there is probably presynaptic dopaminergic dysfunction in Parkinson's disease-affected individuals that precedes Parkinson's disease onset.
Although imaging methods are useful to measure DA dynamics in living human brains, most techniques use indirect measurements of DA levels and none has yet directly measured DA release in early parkinsonism. For example, PET measurements of [11]C-raclopride, which competes with DA to bind to D2 receptors, may indeed report on differences in DA release. However, these measurements fundamentally report on D2 receptor availability, and so may also represent alterations in D2 receptor expression, integrity of postsynaptic projections or loss of DA from degeneration of presynaptic terminals. Likewise, differences in 18F-DOPA measurements, a marker of presynaptic DA storage capacity, could indicate a change in the number of dopaminergic cells, but may also be influenced by alterations in DA metabolism and handling in intact neurons. Further, most of the studies performed on prodromal, preclinical or genetic carriers involve small sample sizes, are not longitudinal and rather rely upon comparisons to a nominal control cohort. Incomplete penetrance of many Parkinson's disease-associated mutations means that truly preclinical subjects cannot be confirmed without long-term follow-up. Therefore, although DA release may be defective before degeneration in Parkinson's disease patients, limitations in technology have made the study of DA release in humans challenging. Animal and human cell-based models of Parkinson's disease have been useful in closing this gap.
Brain. 2023;146(8):3117-3132. © 2023 Oxford University Press